This page was published for Genetics 564 at the University of Wisconsin-Madison
What is Sotos syndrome?
Sotos syndrome, or cerebral gigantism, is a rare genetic developmental disorder characterized by extreme overgrowth in childhood, obscured facial features, and impaired intellectual development [2]. These notable facial features (Figures 1,2) include a bossing of the forehead, prominent jaw, and general pear-shaped face structure to result in a subtly oversized head shape. This facial gestalt is commonly seen to include a lack of hair in the protruding forehead region and a discoloration in the cheeks. Children who have this disease also have significantly advanced bone age in congruence with scoliosis due to the extreme overgrowth during the first six years of life [3]. Additionally, there is a strong incidence of hypotonia that can readily lead to heart defects and a delay in early motor skill development [2].
Clinical findings show variable levels of learning disabilities and behavioral issues in a high percentage of Sotos cases. Reports show a high prevalence of speech delays and an inability to grasp abstract ideas and practical reasoning problems [3]. However, it is key to note that the degree to which these disabilities manifest varies greatly from case to case. For some, slower learning capabilities eventually lead to a fairly average IQ, while others continue to struggle with difficulties throughout adulthood [4].
The most noteworthy aspect of Sotos syndrome is the extreme growth seen in the childhood years. Even from birth, lengths tend to be on the higher end of the spectrum, hovering around two standard deviations above the mean. Those with Sotos are usually much taller in comparison to their peer group, however, by the time these children reach adulthood, their heights average in the normal range [4].
Clinical findings show variable levels of learning disabilities and behavioral issues in a high percentage of Sotos cases. Reports show a high prevalence of speech delays and an inability to grasp abstract ideas and practical reasoning problems [3]. However, it is key to note that the degree to which these disabilities manifest varies greatly from case to case. For some, slower learning capabilities eventually lead to a fairly average IQ, while others continue to struggle with difficulties throughout adulthood [4].
The most noteworthy aspect of Sotos syndrome is the extreme growth seen in the childhood years. Even from birth, lengths tend to be on the higher end of the spectrum, hovering around two standard deviations above the mean. Those with Sotos are usually much taller in comparison to their peer group, however, by the time these children reach adulthood, their heights average in the normal range [4].
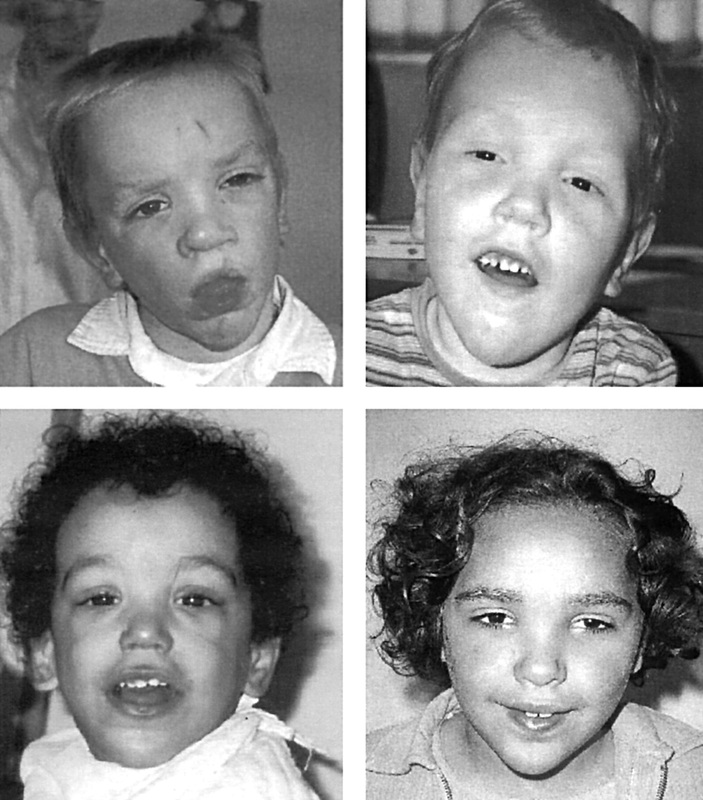
Figure 1: The facial gestalt in childhood, seen by wide set eyes, enlarged forehead, and large head circumference.
|

Figure 2: The facial gestalt in adolescents and adults with Sotos syndrome. Notable features include the protruding chin, enlarged forehead and oversized head.
|
What is the genetic basis?
The gene responsible for the manifestation of Sotos syndrome is the nuclear receptor SET domain-containing protein 1 (NSD1). Located at chromosome 5q35 (Figure 3), this gene encodes for a histone methyltransferase [5]. Localization of NSD1 is spread throughout the body in a number of organs and tissues including the heart, lungs, kidneys, brain, skeletal muscle and spleen [4].
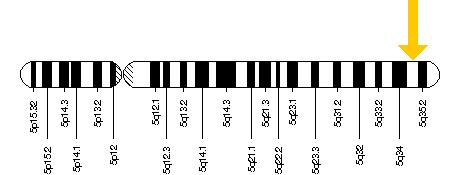
Figure 3: NSD1 is located on the long arm of chromosome 5 at location 35 as depicted by the yellow arrow [6].
NSD1 contains multiple functional domains that help to regulate histone methyltransferase activities of NSD1, although full function is not completely understood [5][6]. Histone methylation is a method of gene regulation that deals with silencing, or "turning off" genes, considering the cellular context. This a key regulating step in development, so it is not surprising that a mutation or deletion at this gene site could illicit such a phenotype. Sotos is most commonly noted by a dominant haploinsufficiency of NSD1, meaning there is only one working copy of the gene in the cells and that one copy is not enough to result in normal function [5]. However, there are over 100 different types of mutations that can lead to Sotos syndrome, including deletions, insertions, and point mutations. Either way, most mutations of NSD1 eventually lead to the inability of one copy of the gene to produce a functioning protein [5][6].
Various studies have found different mutations in a wide range of subjects, although one commonality lies in Japanese patients with Sotos. These patients tend to consistently show 5q35 microdeletions brought about by nonrandom homologous recombination between flanking low-copy number repeats [7]. Outside of Japan, the incidence of this microdeletion is reported in only 10% of cases [4]. The reason for this is still not known.
Various studies have found different mutations in a wide range of subjects, although one commonality lies in Japanese patients with Sotos. These patients tend to consistently show 5q35 microdeletions brought about by nonrandom homologous recombination between flanking low-copy number repeats [7]. Outside of Japan, the incidence of this microdeletion is reported in only 10% of cases [4]. The reason for this is still not known.
How is a person diagnosed?
Diagnosis for Sotos syndrome stems from the existence of the three key clinical features: facial gestalt, childhood overgrowth, and a learning disability. These features are much more apparent during the first six years of life because as the child grows, the severity tends to fade to result in a more subtle appearance. Prior to development of genetic methodologies for Sotos, diagnosis was purely based off of physical features. However, due to high variability in phenotypic manifestation, it can sometimes be quite difficult to obtain a certain diagnosis based solely on these factors. In 2002, researchers isolated a number of genetic mutations consistent with Sotos syndrome, which allowed a more definite diagnosis to be made [8]. So, if a child only exhibits one or two of these features, genetic testing (Figure 4) may be required to observe an abnormality in the NSD1 gene [9].
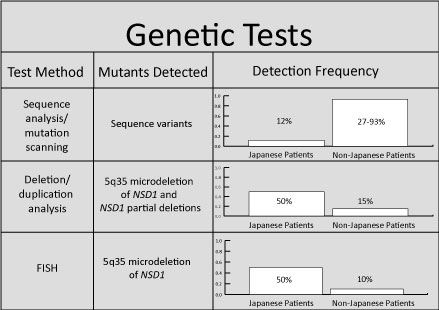
Figure 4: I gathered data to create this chart in order to outline the three genetic tests most commonly used to determine the genetic abnormality. Since Japanese patients with Sotos syndrome have a more specified microdeletion in 5q35, the prevalence was compared to those non-Japanese patients. Sequence analysis is used to identify insertions or deletions, duplication analysis is used to determine if there is a gene duplication, and FISH or fluorescent in situ hybridization is used for chromosomal analysis via labeled DNA probes. [9]
References:
[1] IMAGE: http://www.nature.com/ejhg/journal/v11/n11/fig_tab/5201050f1.html
[2] Genetics Home Reference. (2014). Sotos Syndrome. Retrieved February 3, 2014, from http://ghr.nlm.nih.gov/condition/sotos-syndrome
[3] Cole, TRP., Hughes, HE. (1994). Sotos syndrome: a study of the diagnostic criteria and natural history. Journal of Medical Genetics, 31. 20-32. doi: 10.1136/jmg.31.1.20
[4] Faravelli, F. (2005). NSD1 mutations in Sotos syndrome. American Journal of Medical Genetics, 137C(1), 24-31. doi: 10.1002/ajmg.c.30061
[5] Tatton-Brown, K., Douglas, J., et.al. (2005). Genotype-Phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. The American Journal of Human Genetics, 77(2), 193-204. doi: http://dx.doi.org/10.1086/432082
[6] Genetics Home Reference. (2014). NSD1. Retrieved: February 4, 2014, from http://ghr.nlm.nih.gov/gene/NSD1
[7] Visser, R., Shimokawa, O., et.al. (2005). Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. The American Journal of Human Genetics, 76(1), 52-67. doi: http://dx.doi.org/10.1086/426950
[8] Kurotaki, N., Imaizumi, K., et.al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet, 30(4), 365-366. doi: 10.1038/ng863
[9] Tatton-Brown K., Cole TRP., Rahman N. Sotos Syndrome. (2004) [Updated 2012 Mar 8]. GeneReviews™. http://www.ncbi.nlm.nih.gov/books/NBK1479/
[1] IMAGE: http://www.nature.com/ejhg/journal/v11/n11/fig_tab/5201050f1.html
[2] Genetics Home Reference. (2014). Sotos Syndrome. Retrieved February 3, 2014, from http://ghr.nlm.nih.gov/condition/sotos-syndrome
[3] Cole, TRP., Hughes, HE. (1994). Sotos syndrome: a study of the diagnostic criteria and natural history. Journal of Medical Genetics, 31. 20-32. doi: 10.1136/jmg.31.1.20
[4] Faravelli, F. (2005). NSD1 mutations in Sotos syndrome. American Journal of Medical Genetics, 137C(1), 24-31. doi: 10.1002/ajmg.c.30061
[5] Tatton-Brown, K., Douglas, J., et.al. (2005). Genotype-Phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. The American Journal of Human Genetics, 77(2), 193-204. doi: http://dx.doi.org/10.1086/432082
[6] Genetics Home Reference. (2014). NSD1. Retrieved: February 4, 2014, from http://ghr.nlm.nih.gov/gene/NSD1
[7] Visser, R., Shimokawa, O., et.al. (2005). Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. The American Journal of Human Genetics, 76(1), 52-67. doi: http://dx.doi.org/10.1086/426950
[8] Kurotaki, N., Imaizumi, K., et.al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet, 30(4), 365-366. doi: 10.1038/ng863
[9] Tatton-Brown K., Cole TRP., Rahman N. Sotos Syndrome. (2004) [Updated 2012 Mar 8]. GeneReviews™. http://www.ncbi.nlm.nih.gov/books/NBK1479/
This page was created by Jamie Masliah, an undergraduate at the University of Wisconsin-Madison for Genetics 564.
Last updated: May 24, 2016
